![]() Manual for AIMAll (Version 14.11.23)
Manual for AIMAll (Version 14.11.23)

AIMQB is the "driver" application for setting up and running most QTAIM calculations with AIMAll. AIMQB automatically runs, with various options, a full QTAIM numerical analysis of one or more molecular wavefunction using the programs AIMExt, AIMInt and AIMSum. Results from these programs are summarized in easy-to-read .sum files (a .sum file is a text file with the same base name as the corresponding wavefunction file but with the extension .sum). Results from these programs can also be interactively displayed using AIMStudio. Standard output (such as calculation progress) from AIMQB calculation steps can be displayed in an AIMQB log window or (when running AIMQB from a command-line) in a console window (Windows) or terminal (Mac OS X and Linux).
AIMQB can be run either in Dialog mode (GUI mode) or in Command-Line mode (non-GUI mode).
The general usage statement for AIMQB is: aimqb [options] [wfnfile | wfxfile | fchkfile]
AIMQB can be launched in Dialog mode from Windows, Mac OS X or Linux by double-clicking an AIMQB application icon or by dragging and dropping a wavefunction file onto an AIMQB application icon.
AIMQB can also be launched in Dialog mode from the "Run" menu of AIMStudio.

AIMQB can be launched from a command-line either in Dialog mode or Command-Line mode. A detailed description of using AIMQB with options from a command-line is given here.

Launching AIMQB by double-clicking an AIMQB application icon or from the "Run" menu of AIMStudio or by running aimqb from a command-line (without the -run or -nogui arguments) will display an AIMQB Dialog. An AIMQB Dialog
consists of five tabs:
| Tab | Purpose |
| Wavefunctions | Choose the wavefunction files to use for calculations |
| Atoms | Specify the set of atoms to calculate |
| Methods | Specify the methods and method parameters to use for the calculations |
| Properties | Specify what properties to calculate |
| Other | Miscellaneous |
The "Wavefunctions" tab is shown above while the "Atoms", "Methods", "Properties" and "Other" tabs are shown below.
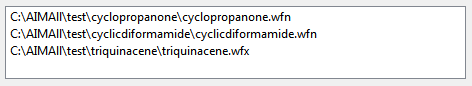
The Wavefunction List contains the file paths of the wavefunctions (.wfn files, .wfx files, .fch files or .fchk files) on which AIMAll calculations are to be done. The Wavefunction List must contain at least one wavefunction file path. If more than one wavefunction file is in the Wavefunction List, the wavefunctions will be processed serially, i.e., in batch mode, beginning with the first wavefunction in the List and ending with the last Wavefunction in the List. Wavefunctions can be added to the List using the "Browse" button or the "Recent" button. Wavefunctions can also be added to the Wavefunction List by dragging the file icons from outside of AIMQB (e.g., from an Explorer window or desktop) and dropping them into the Wavefunction List. Selected wavefunctions can be removed from the Wavefunction List using the Delete keyboard key. The order of the Wavefunctions in the List can be rearranged by dragging and dropping files within the list. The Wavefunction List can be cleared using the "Clear" button. Note that the AIMQB Dialog settings will be used for each of the wavefunctions in the List. If non-default settings are specified, care must be taken to ensure that they are appropriate for each of the wavefunctions in the Wavefunction List.
Note: Running an AIMQB job with a Wavefunction List of more than 3 wavefunction files requires an AIMAll Professional license.

Selecting the "Browse" button will launch a native Windows, Mac OS X or Linux file open dialog for selecting one or more wavefunction files in a directory to be added to the Wavefunction List to run the AIMAll calculations on. To add wavefunction files from multiple directories to the Wavefunction List, just "Browse" multiple times.
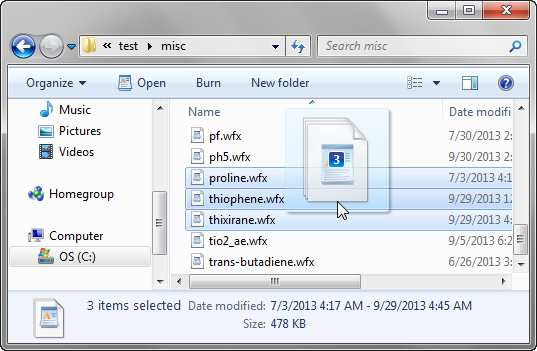
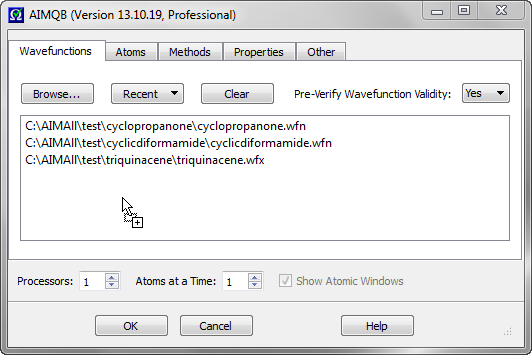

Wavefunction can be added to the Wavefunction List by dragging the file icons from outside of AIMQB (e.g., from an Explorer window or desktop) and dropping them into the Wavefunction List. In the above example, proline.wfx, thiophene.wfx and thixirane.wfx are added to the Wavefunction List by dragging their file icons from a Windows explorer window and dropping them into the List.
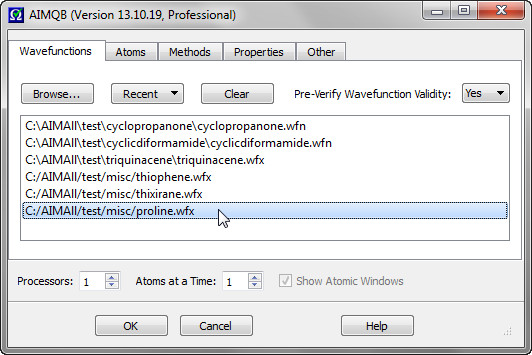
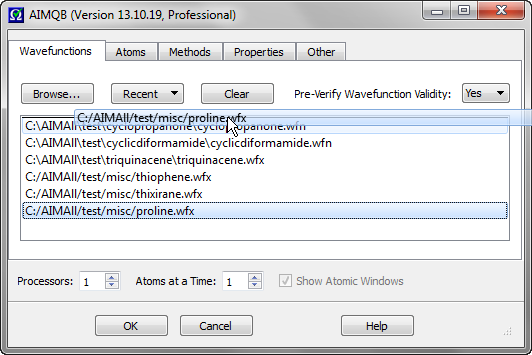
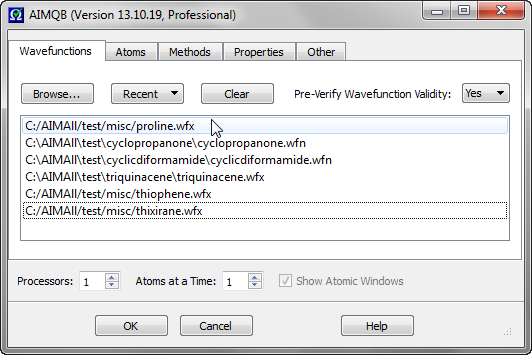
The order of the Wavefunctions in the List can be rearranged by dragging and dropping files within the list. In the above example, proline.wfx is moved from the bottom of the List to the top.
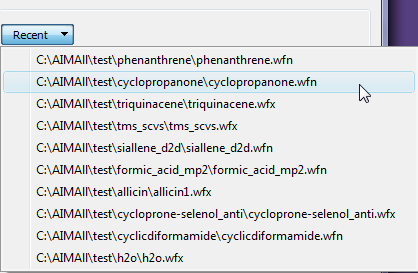
The "Recent" menu presents the 10 most recent wavefunction files that were used with AIMQB. Selecting a wavefunction file from the "Recent" menu will add it to the Wavefunction List.
Clicking the "Clear" button will clear the Wavefunction List.
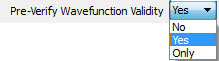
For each AIM wavefunction (.wfn file or .wfx file) in the Wavefunction List, controls whether a verification of the validity of the AIM wavefunction will be done before proceeding with the AIMAll calculations for that wavefunction. Most importantly, an orthonormality check of the MOs of the AIM wavefunction is done. Default is "Yes", meaning the verification will be done and if the wavefunction is not acceptable then an error message will be issued and the AIMAll calculations will not proceed for that wavefunction. Specifying "No" will skip the verification, which is NOT advisable. Specifying "Only" will perform the verification of the wavefunction, inform the user of whether the wavefunction is acceptable or not and then stop, i.e., no calculations will be done even if the wavefunction is acceptable.
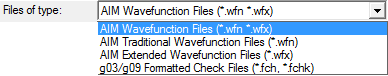
AIMQB accepts three types of wavefunction data files.
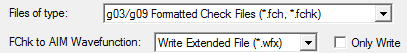
When a g03 or g09 formatted checkpoint file is specified as a wavefunction file, the user has a choice of creating a corresponding .wfx or .wfn file that will be used by the other programs of AIMAll. The default is to create a .wfx file. When "Only Write" is checked, then AIMQB will terminate after the .wfx or .wfn file is created, instead of proceeding with the usual AIMAll calculations.
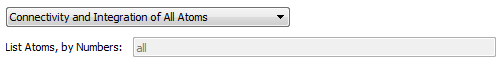
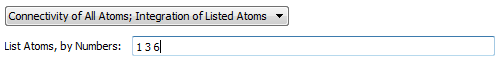
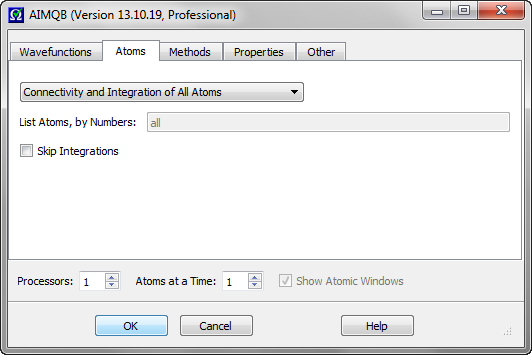
These widgets control which atoms to determine critical point connectivity for and calculate atomic properties for. The default is to determine connectivity and atomic properties of all atoms. The option "Connectivity of All Atoms; Integration of Listed Atoms" will
calculate a full molecular graph but will only calculate atomic properties of the listed atoms. The option "Connectivity and Integration of Listed Atoms" (recommended for reruns of
problem atoms following an all atom run) will only determine the critical point connectivity and atomic properties of the listed atoms, i.e., the full molecular graph will not be (re)calculated. In the last case, if the full
critical point connectivity and other critical point data is already present in a .mgp or .mgpviz file from a previous AIMQB run, then that data will be preserved if the critical points are validated. When listing atoms to calculate,
use a space-separated list of atom numbers. For example, 1 3 6

This checkbox controls whether to skip the atomic integrations. The default is unchecked (false). Note that the atomic integration input files (.inp) for the AIMInt program will still be written even if this box is checked, as will a .bat script (Windows) or .sh script (Linux and Mac OS X) for running the atomic integrations.

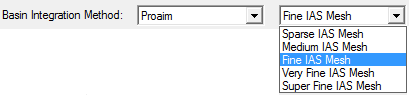

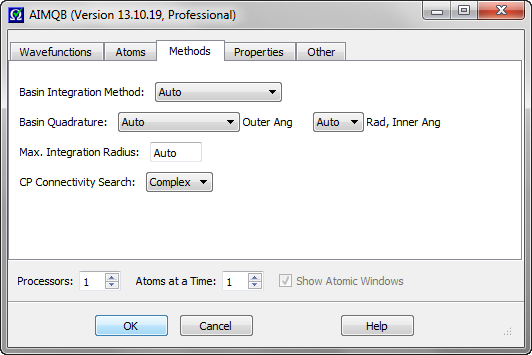
AIMAll supports five different atomic basin integration methods: "Auto", "Proaim", "Promega (1st-order)", "Promega (3rd-order)" and "Sculpt". The "Auto" method is the default, which means AIMAll will automatically decide which of the remaining methods to use to achieve atomic integrations results of good numerical accuracy.
NOTE: As of AIMAll (Version 12.05.09), the default atomic integration method and defaults for atomic integration parameters have been changed to "Auto", which means that AIMAll will automatically determine the appropriate initial atomic integration method and integration parameters for each atom and automatically perform appropriate recalculations of "Problem Atoms" as necessary to achieve atomic integration results of good numerical accuracy. Sometimes, however, advanced users may wish to choose a specific integration method and / or specific integration parameters, in which case the following discussion is of interest.
For AIMAll, much time and effort has been spent on improving the robustness of the default "Proaim" Basin Integration method (compared to its original implementation in AIMPAC) since this method is the most efficient. Development work on further improvement of the "Proaim" algorithm continues, with the goal of making it even more robust.
It is highly recommended that the default "Proaim" Basin Integration method and the default Basin Quadrature parameters always be tried first since they usually work quite well for most atoms and are relatively fast.
Only the "Proaim" method can be used to calculate interatomic surface properties.
In the event that integration results for one or more atoms are not sufficiently accurate (or are unavailable) using the default integration options, you can rerun AIMQB for just those problem atoms, using different integration options along with the "Atoms to Calculate" fields near the bottom of the AIMQB dialog.
If an atom has a "potentially significant" but not "significant" or serious integration error, simply increasing the "Outer Ang" Basin Quadrature to "Very High" or "Sky High" while still using the "Proaim" Basin Integration method might help, and is not too expensive.
The "IAS Mesh" options are specific to the "Proaim" method and control the maximum allowed spacing between adjacent interatomic surface (IAS) paths and hence the fineness of the IAS triangulation and the accuracy of the intersections of the basin integration rays with the IASs. The default "Fine IAS Mesh" is usually quite sufficient for accurate atomic integrations but the "Very Fine" or "Super Fine" IAS Meshes in conjunction with large quadratures may sometimes be useful for very accurate atomic integrations.
Using the "Promega (1st-Order)" basin integration method is usually a good choice for atoms whose integration results are significantly in error (or are unavailable due to a failure to connect a BCP to an RCP) using the default "Proaim" method. Note, however, that the "Promega (1st-Order)" method is significantly more expensive than the "Proaim" method.
The "Promega (3rd-Order)" method is similar to the "Promega (1st-Order)" method except that up to 3 intersections of the integrations rays with the atomic surfaces are searched for. While the "Promega (3rd-Order)" is at least as accurate and sometimes more accurate than the "Promega (1st-Order)" method, it is also slower.
The method of last resort is usually the "Sculpt" method, which is very expensive but will always work in the rare event that the other methods fail to deliver acceptable accuracy. The non-default "Extended Capture" option can be used to significantly speed up the "Sculpt" method, at the slight risk of causing the method to fail.
Once a rerun of AIMQB for just the specified problem atoms is done, the sumfile will automatically be properly upated to include the results for the newly calculated atoms.
Note that for molecules with very diffuse charge distributions, such as some anions or excited states, increasing the maximum atomic integration radius may be necessary to obtain good accuracy, independent of the basin integration method and basin quadrature parameters. This section assumes that the maximum atomic integration radius has been set sufficiently large.
NOTE: As of AIMAll (Version 12.05.09), the default atomic integration method and defaults for atomic integration parameters have been changed to "Auto", which means that AIMAll will automatically determine the appropriate initial atomic integration method and integration parameters for each atom and automatically perform appropriate recalculations of "Problem Atoms" as necessary to achieve atomic integration results of good numerical accuracy. Sometimes, however, advanced users may wish to choose a specific integration method and / or specific integration parameters, in which case the following discussion is of interest.
Atomic basin properties are calculated using 3D numerical integration. For the purposes of the 3D integration, the atom is broken up into two major regions: an "Inner" region that is a sphere (Beta sphere) centered on the nucleus and contained within the atom; and an "Outer" region that is the rest of the atom. Both the Inner region and Outer region integrals are done using a combination of radial and angular numerical integration quadratures.
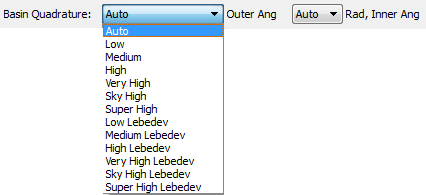
The most important Basin Quadrature control is the "Outer Ang" control, which determines the angular quadrature to be used for the "Outer" region of the atom. The first six entries in the "Outer Ang" combobox (Low, Medium, High, Very High, Sky High and Super High) correspond to uniform and Gauss-Legendre quadratures for the Phi and Theta spherical polar angular coordinates, respectively. The last six entries of the "Outer Ang" combobox (Low Lebedev to Super High Lebedev) use Lebedev quadratures for the angular integration of the Outer region of the atom. The default "Outer Ang" quadrature is "Very High Lebedev". The largest and most expensive "Outer Ang" quadrature is "Super High". The larger grids may be useful for integrating atoms with very complex (but still accurately determinable) surface shapes. The Lebedev quadratures are especially useful for the very expensive Vee(A,A) and Vee(A,B) calculations.
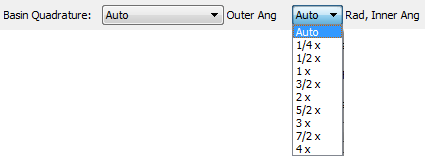
The radial quadrature of both the Outer and Inner regions of the atom and the angular quadrature of the Inner region of the atom are controlled using the "Rad, Inner Ang" combobox. The entries in the "Rad, Inner Ang" combobox correspond to multiples of the default ("1 x") quadrature. The "1 x" radial and inner angular quadrature is almost always recommended and sufficient for calculation of most atomic properties, and certainly "3/2 x" is. A possible exception is atomic volume properties, which need higher radial quadrature for the Outer region of the atom to achieve the same level of accuracy as most other atomic properties calculated at a lower radial quadrature. This is because the isosurfaces that "cap" the atomic volumes are not determined explicitly by AIMInt. The "Auto" radial and inner angular quadrature is essential for Vee(A), Vee(A,A), Vee(A,A'), Vee(A,B) and calculations of atomic Ehrenfest forces as -<Psi|Grad1V|Psi>(A), for which it is the default.
As of AIMAll (Version 14.06.21), the "Rad, Inner Ang" Basin Quadrature control has been disabled, with the "Auto" setting always being used.
Note that for molecules with very diffuse charge distributions, such as some anions or excited states, increasing the maximum atomic integration radius may be necessary to obtain good accuracy, independent of the basin integration method and basin quadrature parameters. This section assumes that the maximum atomic integration radius has been set sufficiently large.
NOTE: As of AIMAll (Version 12.05.09), the default atomic integration method and defaults for atomic integration parameters have been changed to "Auto", which means that AIMAll will automatically determine the appropriate initial atomic integration method and integration parameters for each atom and automatically perform appropriate recalculations of "Problem Atoms" as necessary to achieve atomic integration results of good numerical accuracy. Sometimes, however, advanced users may wish to choose a specific integration method and / or specific integration parameters, in which case the following discussion is of interest.

The maximum atomic integration radius editor control determines the maximum radial boundary, in atomic units, that can be used for the radial integrations of the Outer region of an atom. The default is 13.0. Larger values may sometimes be necessary for some anions or excited states. Any number between 7.0 and 20.0 is allowed.

This combobox controls the intensity of the search for connectivity between the electron density critical points (CPs) found, especially connectivity
between bond critical points (BCPs) and ring critical points (RCPs). "Complex" is the default and searches for all CP connectivities using a very thorough
procedure that can be expensive for systems with many atoms and many RCPs. "Simple" searches for all CP connectivities using a somewhat less thorough and much less expensive
procedure than that corresponding to "Complex". "Basic" searches only for BCP connections to nuclear attractors critical points (NACPs) and non-nuclear
attractor critical points (NNACPs), i.e., bond paths. "Basic" is relatively fast in all cases but is not available when the "Proaim" basin
integration method is used.
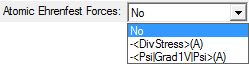
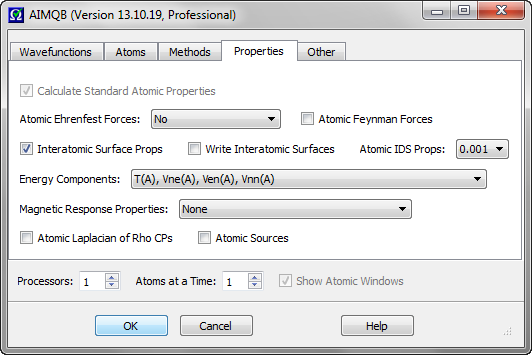
This combobox controls whether and how to calculate atomic Ehrenfest forces. The default is not to calculate any atomic Ehrenfest forces. -<DivStress>(A) means the atomic Ehrenfest forces will be calculated in terms of minus the divergence of the stress tensor. -<Psi|Grad1V|Psi>(A) means the atomic Ehrenfest forces will be calculated in terms of the one-electron gradient of the potential energy operator, which is very expensive (more expensive than Vee(A) calculations). -<DivStress>(A) will also be calculated when -<Psi|Grad1V|Psi>(A) is selected.

This checkbox controls whether to print atomic contributions to the Feynman electrostatic forces on each of the nuclei in the molecule to the .sum and .sumviz files. The default is unchecked (false). Atomic Feynman force data can be quite large (O(N**2)) and users uninterested in this data should consider leaving this box unchecked to avoid cluttering the .int, .sum and .sumviz files.

This checkbox controls whether to calculate interatomic surface (IAS) properties such as IAS areas (out to the 0.0004, 0.001 and 0.002 isodensity surfaces), the number of electrons per unit length, the kinetic energy per unit length, etc. The default is checked (true). This option can only be used in conjunction with the default "Proaim" atomic basin integration method. To calculate the "Surface Virial" energy and the surface "pressure" (surface integral of the Ehrenfest Force density) for each of the IASs, check this box and also select the -<DivStress>(A) option or the -<Psi|Grad1V|Psi>(A) option for "Atomic Ehrenfest Forces".
This checkbox controls whether to write interatomic surface (IAS) data (both GradRho paths and IAS / integration ray intersection data) to atomic .iasviz files, primarily for use with the AIMStudio visualization program. Default is unchecked (false). For a wavefunction file named abc.wfx, the .iasviz files will be written to the abc_atomicfiles subdirectory of the directory containing abc.wfx. These .iasviz files can be quite large (several MB per IAS) and, for large molecules, calculations using this option should typically only be done for a subset of the atoms in the molecule.
Note: Multiprocessor calculation features and all visualization-related features, such as writing .iasviz files and displaying their data in AIMStudio, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
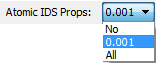
This combobox controls the calculation of atomic isodensity surface (IDS) properties - particularly electrostatic potential (ESP) properties, which can be expensive. The default is "0.001", which means calculate ESP properties and other properties on the atomic portions of the molecule's 0.001 a.u. isodensity surface. Selecting "No" means that ESP properties will not be calculated on any isodensity surfaces. Selecting "All" means calculate ESP properties and other properties on the atomic portions of the molecule's 0.0004, 0.001 and 0.002 a.u. isodensity surfaces. Note that the atomic isodensity surface areas are always calculated.
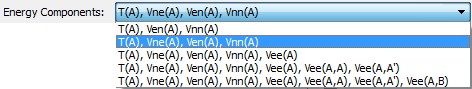
This combobox controls which atomic energy components to calculate. The energy components are as follows:
AIMAll supports two QTAIM total atomic energy definitions: The traditional "Virial-Based" total atomic energy and the newer "Interacting Quantum Atoms (IQA)" total atomic energy based on the work of Pendas et al.
The "Virial-Based" total atomic energy of atom A is defined (by analogy with the molecular total energy expression from the molecular virial theorem) as:
This combobox controls which method, if any, to use to calculate atomic magnetic response properties such as atomic net currents, atomic contributions to NMR shielding tensors and atomic contributions to magnetizabilities. The default is not to calculate atomic magnetic response properties. The CSGTB and IGAIM methods requires a CSGT-based magnetic wavefunction file. The GIAO method requires a GIAO-based magnetic wavefunction file or a .fch(k) file with GIAO based density matrix data.
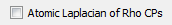
This checkbox controls whether to locate and characterize critical points of the Laplacian of Rho (DelSqRho), where Rho is the electron density. The default is unchecked (false). The Laplacian of Rho critical point data is determined an atom at a time and the data is written to atomic result visualization files ending with the extension .agpviz. For a wavefunction file named abc.wfx, the .agpviz files will be written to the abc_atomicfiles subdirectory of the directory containing abc.wfx. These .agpviz files can be opened in AIMStudio in order to visualize Laplacian of Rho critical points (and corresponding atomic graph gradient paths) and their properties in 3D windows and in interactive Tables.
Note that calculation of Laplacian of Rho critical point data typically significantly increases atomic computation times and users uninterested in this data should avoid checking this box.
Note that in AIMAll, topological analyses of the actual Laplacian of Rho (DelSqRho) are reported (as opposed to the more traditional reporting of minus the Laplacian of Rho (-DelSqRho) analyses). This is done to be consistent with the use of the Laplacian of Rho in other areas of AIMAll (plotting, for example).
The signs of the Hessian of DelSqRho and its eigenvalues are opposite to the signs of the Hessian of -DelSqRho and its eigenvalues. The signatures of critical points of DelSqRho are thus opposite in sign to those of -DelSqRho.Note: Multiprocessor calculation features and all visualization-related features, such as writing .agpviz files and displaying their data in AIMStudio, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.

This checkbox controls whether to calculate and print atomic source contributions to the electron density at electron density critical points and nuclei. The default is unchecked (false). Atomic source data can be quite large (O(N**2)) and users uninterested in this data should consider leaving this box unchecked to avoid cluttering the .int, .sum and .sumviz files.

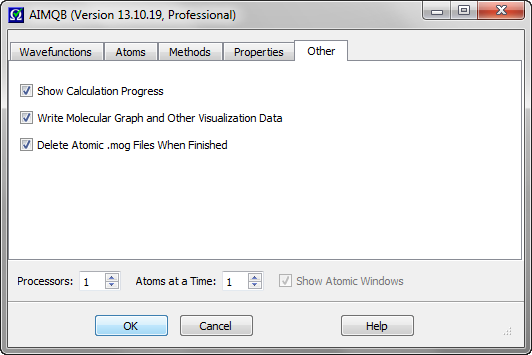
This checkbox controls whether or not to show detailed calculation progress (especially atomic integration progress) in the AIMQB log window. Default is checked.

This checkbox controls whether to write molecular graph and other visualization data to a .mgpviz file (which also contains the same critical point data as the *.mgp file) and a .sumviz file for use with the AIMStudio visualization program. Default is checked.
Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.

This checkbox controls whether to delete the atomic .mog files used for Vee(A,A) and Vee(A,B) calculations when AIMQB is finished with the corresponding wavefunction. Default is checked (true). In some cases,
users may wish to save the atomic .mog files to run additional Vee(A,B) calculations later, in which case this box should be unchecked.

The number of processors (shared memory only) to use for AIMExt and AIMInt jobs. Default is 1. Must be less than or equal to the number of logical processors of the system. If more than one atom at a time is
calculated, then the number of processors to use per atom will be (number of processors)/(number of atoms at a time). Note that the most effective use of multiple processors for small wavefunctions is to calculate more than one atom
at a time (e.g., Processors = 3 and Atoms at a Time = 3). For large wavefunctions, using more than one processor per atomic calculation becomes effective (e.g., Processors = 3 and Atoms at a Time = 1).
Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
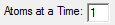
The number of atoms to calculate at a time. Must be less than or equal to the number of processors to use. If the number of atoms to calculate at a time is less than
the number of processors to use, then the number of processors to use per atom will be (number of processors)/(number of atoms at a time). For example,
if 4 processors and 2 atoms at a time are specified (and you have at least a 4 processor system), then two AIMInt jobs will be run at a time, each using 2 processors. Note that the most effective use of multiple processors for small wavefunctions is to calculate more than one atom
at a time (e.g., Processors = 3 and Atoms at a Time = 3). For large wavefunctions, using more than one processor per atomic calculation becomes effective (e.g., Processors = 3 and Atoms at a Time = 1).
Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.

When more than one atom is calculated at a time the main AIMQB log window, console window or terminal will only display a summary of the progress of the atomic
integrations. The "Show Atomic Windows" checkbox controls whether separate log windows should be shown for each atomic
integration calculation. The default is checked. Note that atomic log windows will not be shown if -console or -nogui is specified.
Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
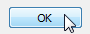
Selecting the "OK" button will close the AIMQB dialog and start the AIMAll calculations on the wavefunction files in the Wavefunction List, the calculation progress being shown in an AIMQB log window by default. If the Wavefunction List contains more than one wavefunction, the wavefunctions will processed serially, i.e., one at a time.
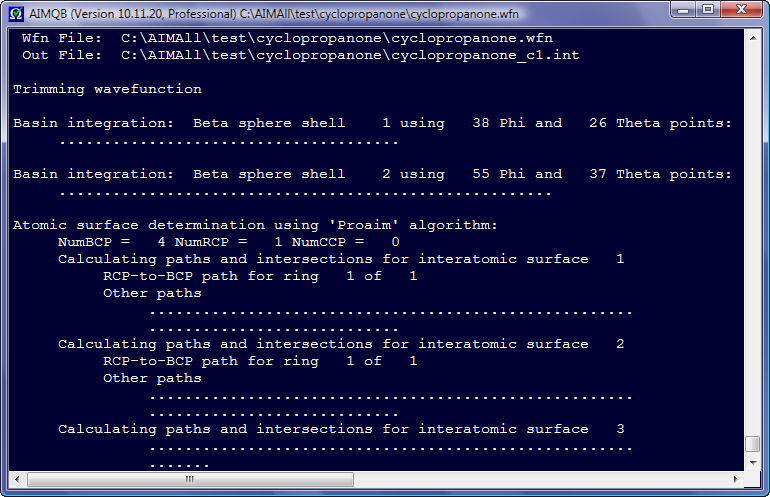
After an AIMQB job is started, an AIMQB log window is shown by default and the progress of the AIMAll calculations for each wavefunction in the Wavefunction List is continuously written to this log window. If you close an AIMQB log window while the AIMQB job is still running, you will be given the option of killing the AIMQB job or leaving it running without the log window.
Right-clicking in an AIMQB log window displays a context menu with options for controlling the appearance of the log window and other self-explanatory items.
Note: Multiprocessor calculation features and all visualization-related features, such as writing .iasviz files and displaying their data in AIMStudio, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
-atidsprops=no/0.001/allNote that calculation of Laplacian of Rho critical point data typically significantly increases atomic computation times and users uninterested in this data should avoid specifying -atlaprhocps=true.
Note that in AIMAll, topological analyses of the actual Laplacian of Rho (DelSqRho) are reported (as opposed to the more traditional reporting of minus the Laplacian of Rho (-DelSqRho) analyses). This is done to be consistent with the use of the Laplacian of Rho in other areas of AIMAll (plotting, for example).
The signs of the Hessian of DelSqRho and its eigenvalues are opposite to the signs of the Hessian of -DelSqRho and its eigenvalues. The signatures of critical points of DelSqRho are thus opposite in sign to those of -DelSqRho.Note: Multiprocessor calculation features and all visualization-related features, such as writing .agpviz files and displaying their data in AIMStudio, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
-wsp=true/falseNote: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
-nproc=1/2/etc.Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
-naat=1/2/etc.Note: Multiprocessor calculation features and all visualization-related features, such as writing and displaying molecular graphs, require an AIMAll Professional license for wavefunctions with more than 12 atoms or more than 400 primitive basis functions.
-shm_lmax=-1/0/1/2/etc.Note that for AIMQB command-line calculations on non-small wavefunctions it is recommended to run AIMQB in the background and either use -scp=false or redirect the standard output to a log file:
/home/todd/AIMAll/aimqb.ish -nogui -scp=false dma.wfx &