 Manual for AIMAll (Version 14.11.23)
Manual for AIMAll (Version 14.11.23)
Tutorial 1 - Getting Started
In this tutorial, we will do a basic QTAIM computational analysis and introductory visual QTAIM analysis of cyclopropanone.wfn
- Launch AIMStudio
 from the desktop or quicklaunch area.
from the desktop or quicklaunch area.
- Select the "Run->AIMQB" menu item to launch an AIMQB Dialog.
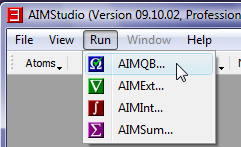
- In the AIMQB Dialog, click the "Browse" button, navigate to the folder C:\AIMAll\test\cylcopropanone (on Mac OS X or Linux the directory would be, for example, ~/AIMAll/test/cyclopropanone) and select the cyclopropanone.wfn file.
- In the AIMQB dialog, click the "OK" button to start an AIMAll numerical analysis of cyclopropanone.wfn
- Wait a minute or two until the AIMQB log window says "Job Completed" then close the log window.
- Select the "File->Open in New Window" menu item and open the file cyclopropanone.sumviz
- Practice manipulating the molecule position and orientation in the window.
- Rotate the molecule about the screen Y-axis by moving the mouse cursor left and right in the window with the left mouse button held down.
- Rotate the molecule about the screen Y-axis by using the left and right arrow keyboard keys.
- Rotate the molecule about the screen X-axis by moving the mouse cursor back and forth in the window with the left mouse button held down.
- Rotate the molecule about the screen X-axis by using the up and down arrow keyboard keys.
- Rotate the molecule about the screen Z-axis by moving the mouse cursor left and right in the window with the right mouse button held down.
- Rotate the molecule about the screen Z-axis by using the Page Up and Page Down keyboard keys.
- Zoom the molecule in and out by using the mouse scroll wheel or the touch pad or magic mouse scrolling mechanism.
- Zoom the molecule in and out using F5 and F6 keyboard keys.
- Move the molecule along the screen X-axis by moving the mouse cursor left and right in the window with the middle mouse button (or scroll wheel) held down.
- Move the molecule along the screen X-axis by moving the mouse cursor left and right in the window with the left mouse button held down and the Shift key pressed.
- Move the molecule along the screen X-axis by using the left and right arrow keyboard keys with the Shift key pressed.
- Move the molecule along the screen Y-axis by moving the mouse cursor up and down in the window with the middle mouse button (or scroll wheel) held down.
- Move the molecule along the screen Y-axis by moving the mouse cursor up and down in the window with the left mouse button held down and the Shift key pressed.
- Move the molecule along the screen Y-axis by using the up and down arrow keyboard keys with the Shift key pressed.
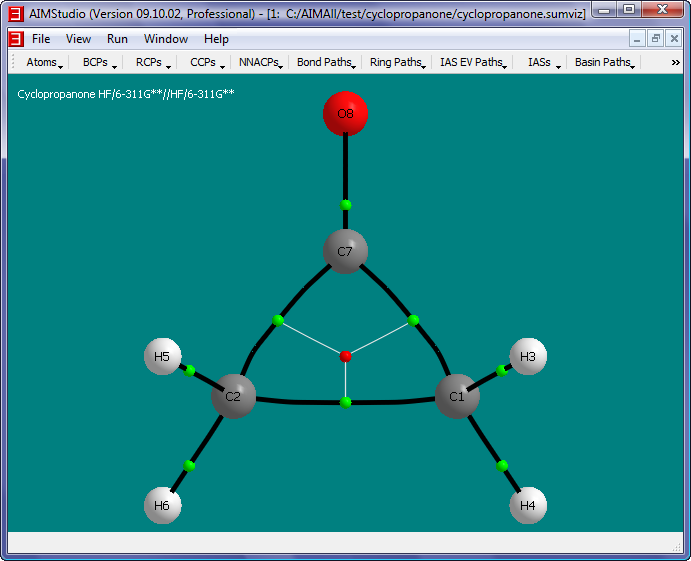
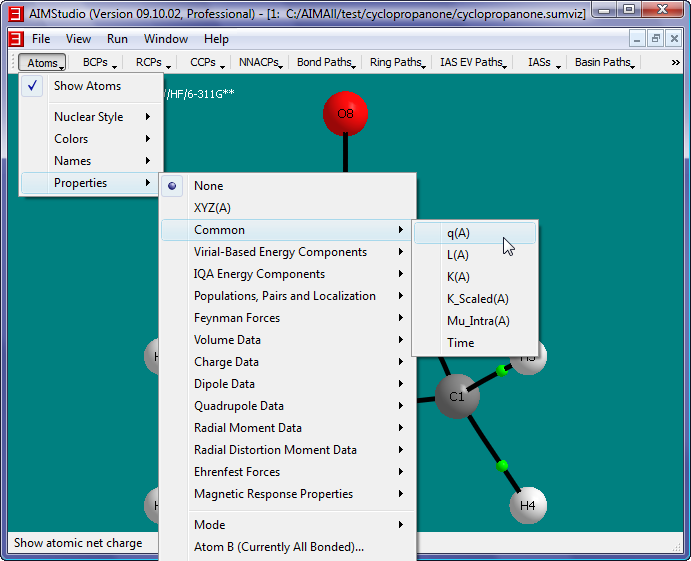
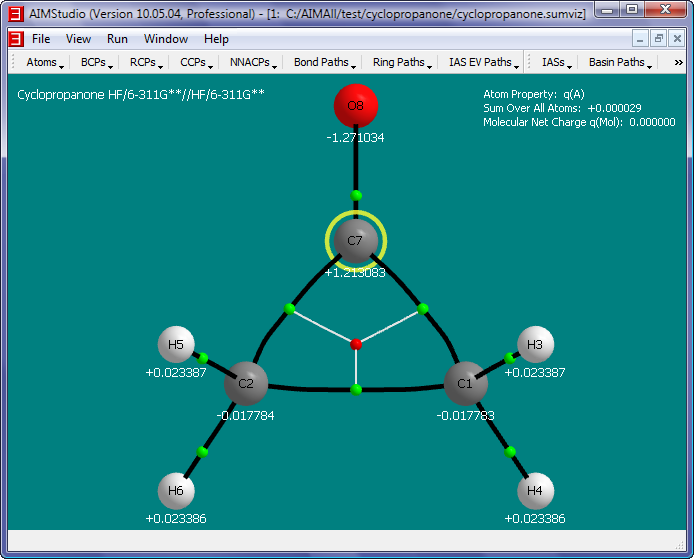
- Show the atomic charges q(A) in the window by checking the menu item "Atoms->Properties->Common->q(A)".
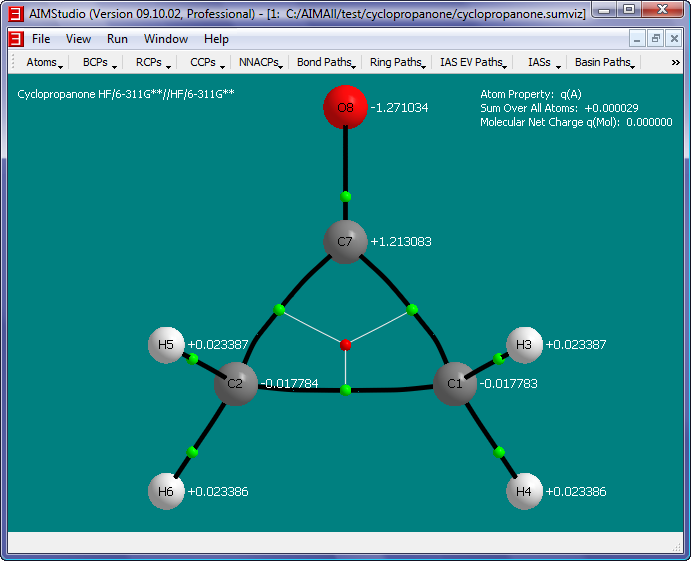
- Place the atom property text below the nuclear spheres (instead of to the right) by checking the "Atoms->Properties->Text->Placement->Bottom" menu item.

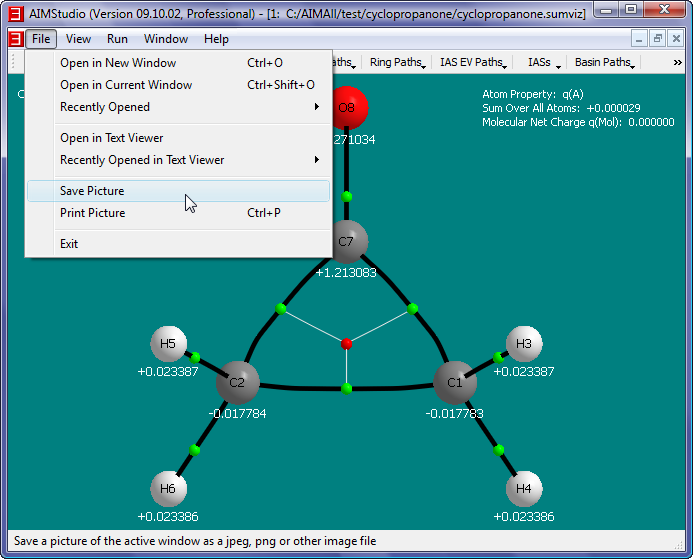
- Save a picture file (named cyclopropanone.png) of the window interior by selecting the "File->Save Picture" menu item.
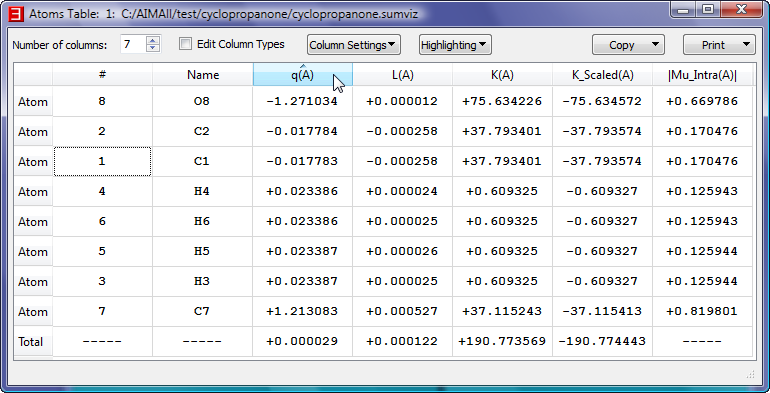
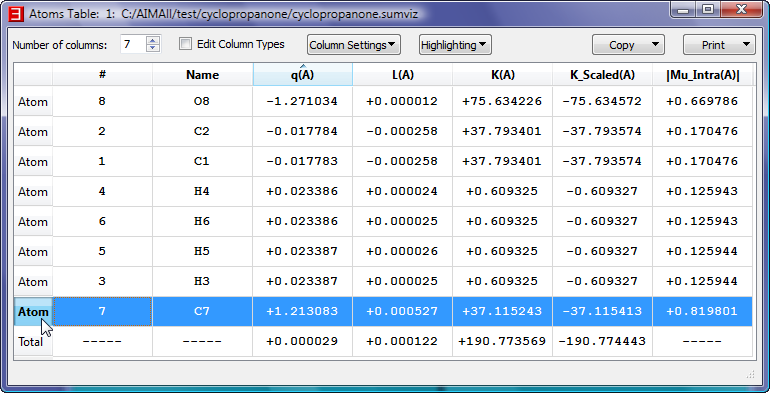
- Show the interactive "Atoms Table" for cyclopropanone.sumviz by selecting "Atoms->Table". In the Atoms Table, click on the atomic charges header "q(A)"
to sort the atoms in ascending order by atomic charge value. Select a cell in the last Atom row (C7) and note that the C7 atom's nuclear sphere is highlighted in the 3D window.
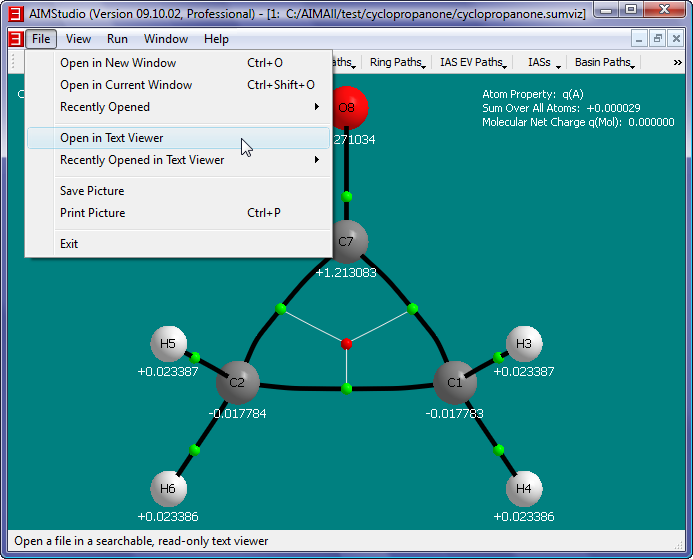
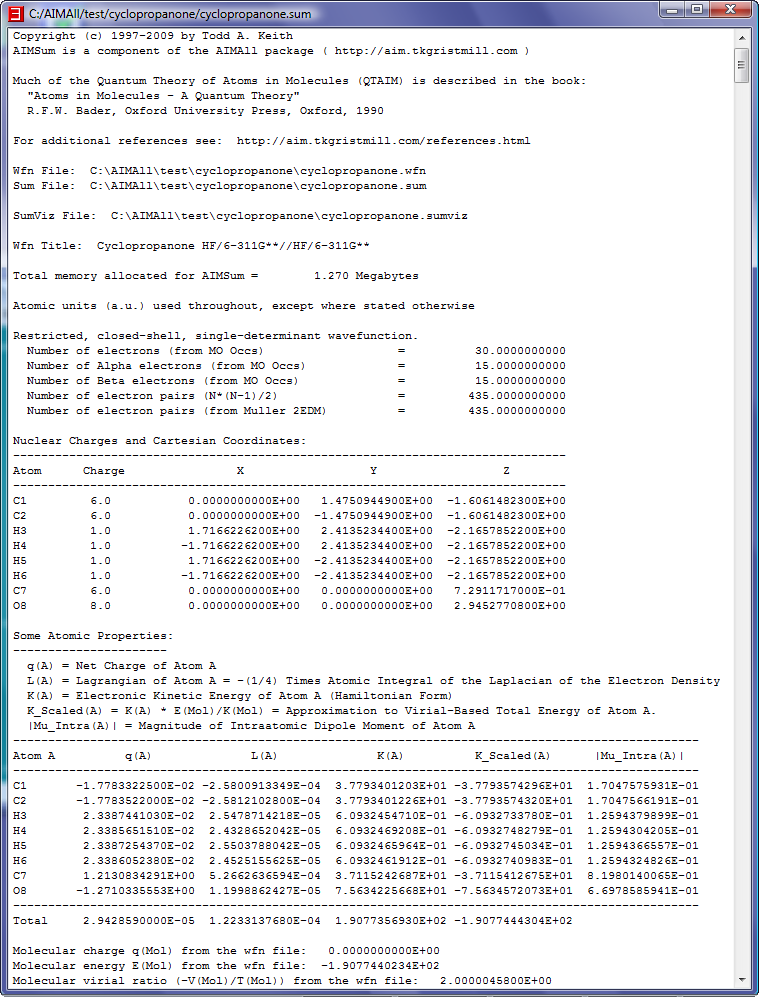
- Look at the full results of the cyclopropanone.wfn analysis in the text result file named cyclopropanone.sum (corresponding to the visualization result file cyclopropanone.sumviz) by selecting
the menu item "File->Open in Text Viewer". Right-click in the text viewer window to popup a menu. Select the "Find..." menu item to show a "Find Text" dialog.
Copyright © by Todd A. Keith, 1997-2014 (aim@tkgristmill.com)